








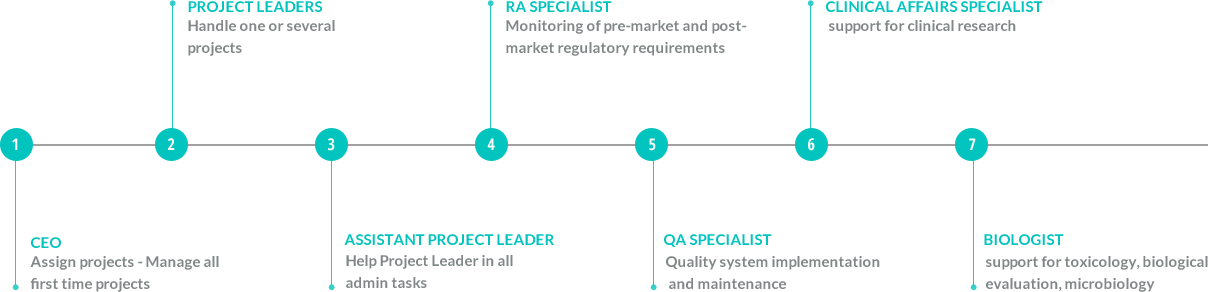
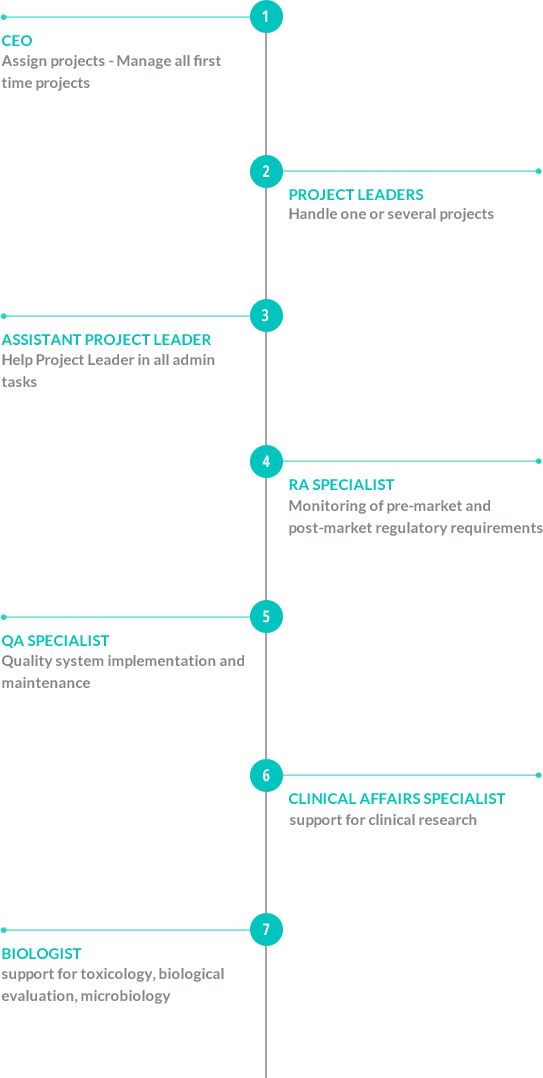





Medical Device Classes
Introduction United States Food and Drug Administration (FDA) regulates all medical devices manufactured in the U.S. The FDA regulations, coupled with European Commission regulations, nearly cover all international distribution of…

